Diagnosis & Treatment of Phenylketonuria (PKU)
Phenylketonuria
is an inborn error of metabolism
characterized by high levels of the amino acid phenylalanine [Phe] in the blood of
newborns. PKU is
diagnosed by means of various versions of the Guthrie Test, first
devised by Dr Robert Guthrie (1916 - 1995) after the
birth of his child with PKU.
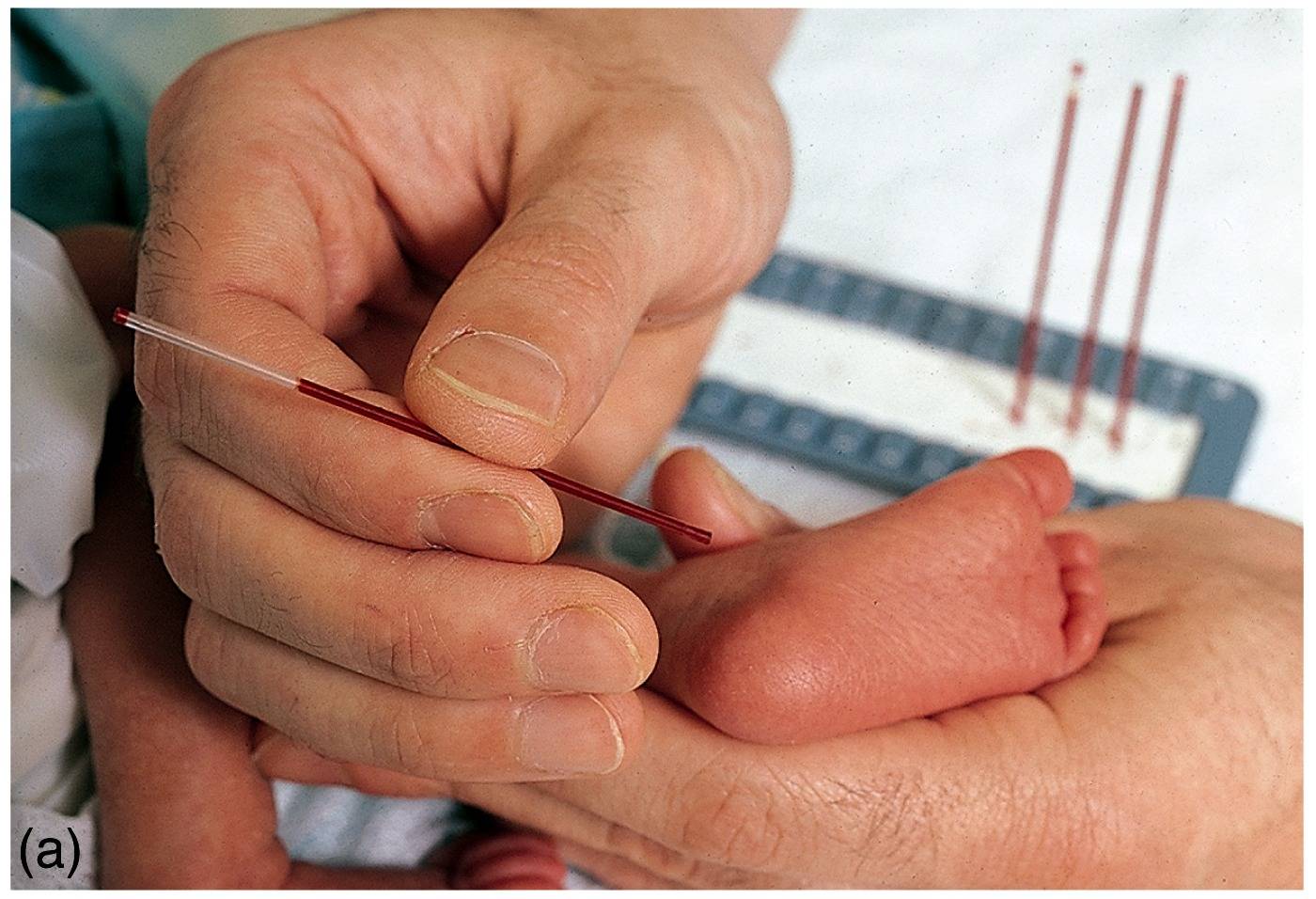
In the original test, a small drop of blood taken from the
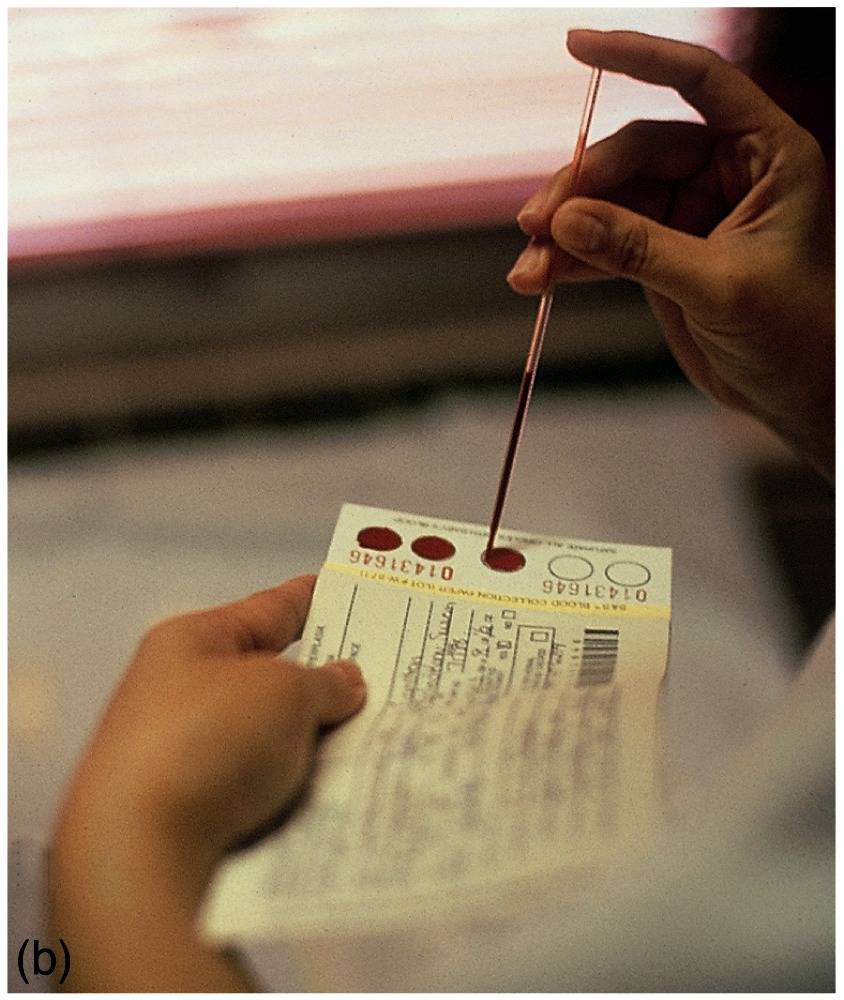
heel of a newborn (left) was applied to a filter-paper disc
[right]. A punch-out of the dried disc was incubated on a
Petri dish plated with bacteria (Bacillus subtilis), in the presence of a
growth inhibitor, B-2-thienyl-alanine.
High levels of Phe in the blood sample
overcame the inhibition, and allowed bacterial growth in the
area around the disc. The original test
has been supplanted by direct mass-spectroscopy measurement of
Phe concentration in
the blood sample. Conceptions at risk for PKU may also be
screened by prenatal PCR-based
DNA tests for the PKU-related alleles
themselves.
If an elevated level of Phe is detected, the child is placed on a phenylalanine-restricted diet for their first several years (< 10 years). This can completely offset the expected neurological effects (below). The Guthrie Test was the first widely-used peri-natal biochemical diagnostic test. Since its introduction in the early 1960s, it been used in >90% of North American hospitals, and in most other countries. Guthrie's method has been extended to many other conditions.
Current evidence suggests that high levels of [Phe] may impair mental performance later in life, and that a Phe-restricted diet should be maintained throughout life.
 .
.
If an elevated level of Phe is detected, the child is placed on a phenylalanine-restricted diet for their first several years (< 10 years). This can completely offset the expected neurological effects (below). The Guthrie Test was the first widely-used peri-natal biochemical diagnostic test. Since its introduction in the early 1960s, it been used in >90% of North American hospitals, and in most other countries. Guthrie's method has been extended to many other conditions.
Current evidence suggests that high levels of [Phe] may impair mental performance later in life, and that a Phe-restricted diet should be maintained throughout life.
.