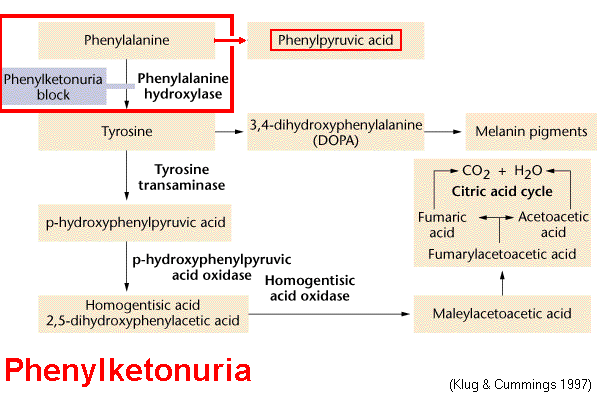
Inborn errors of Phenylalanine metabolism: Phenylketonuria
Phenylketonuria
(PKU) is
a metabolic disease characterized by accumulation of Phenylalanine
(Phe) and its
by-products in the
blood of fetuses and
newborns. PKU results from a defect in the enzyme Phenylalanine Hydroxylase (PAH), such that Phe
is not converted to Tyrosine. The alternative
conversion
of Phe to PPA causes accumulation of
the
latter as a toxin in
the
Central Nervous System, which can lead to severe mental
retardation.