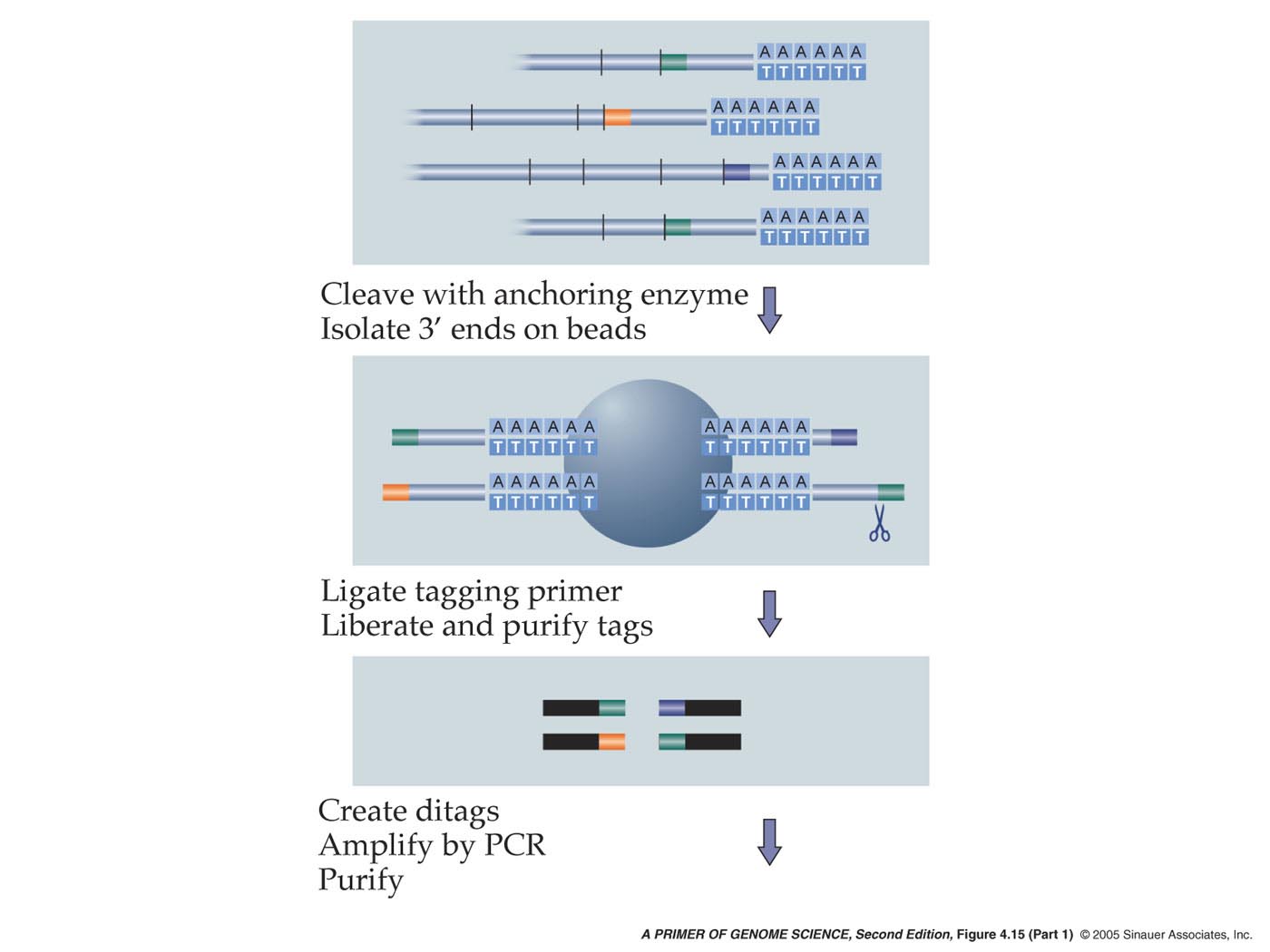
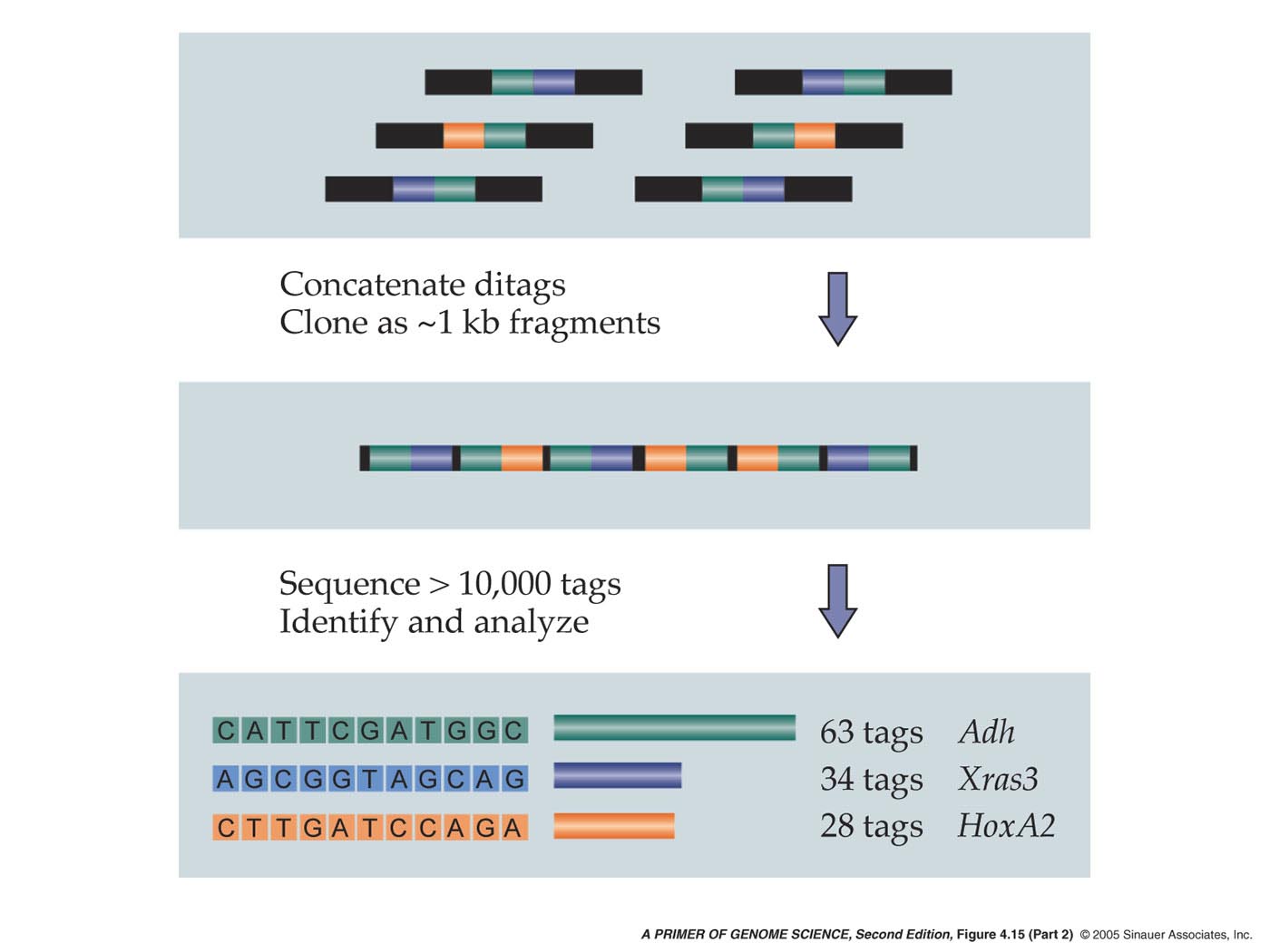
-SAGE is a method for determining the absolute abundance of every transcript expressed in a population of cells through the sequencing of 15 bp tags unique to every gene.
This method of analysing the transcriptome begins by constructing a double-stranded cDNA library from polyA mRNA and then cleaving it with an anchor enzyme usually NlaIII, which typically cut every 400 bases. The polyA tail is then bound to magnetic beads coated with Streptavidin. Once bound, another restriction enzyme cleaves off the tags plus an additional 15 base pairs. These fragments are ligated together in pairs to form ditags, which are then PCR-amplified, using primers complementary to the 15 base pair adapters. After amplification, the adapters are cleaved off and all the 30 base pair ditags are ligated together, producing approximately 1-kb fragments, which are subsequently inserted into a vector, cloned and sequenced. A list of each tag and its abundance is compiled and the gene associated with each tag is identified.
Transcript abundance can be either expressed in relative terms or as the number of transcripts per cell. This method is advantageous because it is unbiased by factors such as reference samples, hybridization artifacts or clone representation and it yields an accurate representation of the true number of transcripts per cell. An important drawback is that the high cost makes replicates and large number of samples difficult.