"Forward" Mendelian Genetics versus "Reverse" Molecular Genetics:
Genetics and Molecular Biology of Alkaptonuria, an inborn error of metabolism
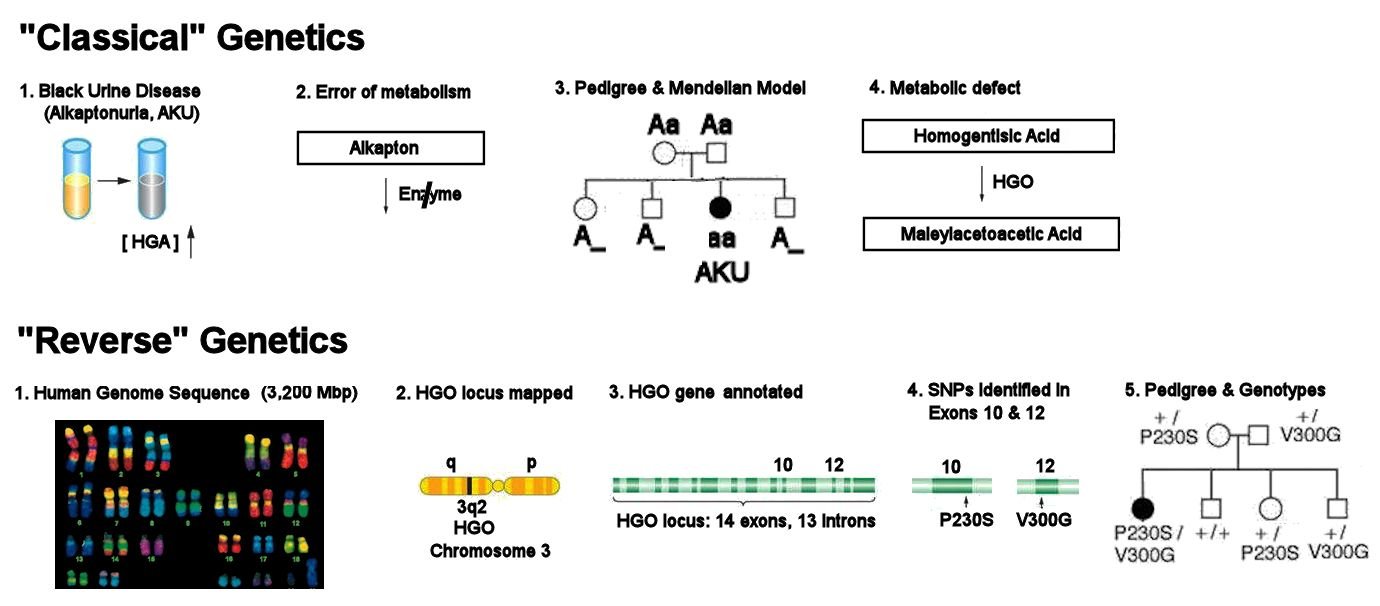
In the first classical genetics approach to a human
trait, the physician Archibald Garrod in 1902
observed "Black Urine
Disease" (Alkaptonuria, AKU) in
his patients (Step
1). The urine
and diapers of infants with Alkaptonuria
darken upon exposure to air, and adults show darkening of
the cartilage in the ears and nose. Chemical analysis
identified a high level of a substance called alkapton in their urine
(Step 2). From the pattern of inheritance (pedigree) observed in
families under his care (two unaffected parents, and
unaffected and affected children in an approximate 3:1
ratio) (Step
3), Garrod deduced that the condition was inherited as a recessive trait, following
the reasoning of Gregor
Mendel whose work in 1867 had recently been
rediscovered, and was widely discussed. Following Mendelian Rules,
the birth of an alkaptonuric child to two unaffected parents
suggests that she had two recessive alleles (aa) at some Gene for the trait. The
parents must both then be Aa, and do not show the
condition because A is dominant to a.
The other, unaffected children are either AA or Aa
(shown as A-). Garrod further suggested that the
condition was due to the absence of an enzyme to
metabolize (break down) alkapton. Subsequent biochemical
analysis showed that alkapton (now called Homogentisic
Acid) is metabolized by Homogentisic Acid Oxidase
(HGO) to Maleylacetoacetic Acid (Step 4). Thus, genetic
analysis of crosses in a pedigree allows inference of the
existence and recessive nature of a gene for the trait
Alkaptonuria. The physical nature of the gene was at that
time entirely unknown.
Molecular Genetics in the 21st century proceeds from knowledge of the 3,200 Mbp human genome, completed in 2003 (Step 1). Based on knowledge of the amino acid sequences of HGO in other organisms, bioinformatic analysis of all 23 pairs of chromosomes mapped the gene for Homogentisic Acid Oxidase (HGO) to Band 2 on the long (q) arm of Chromosome 3 (3q2) (Step 2). Detailed sequence analysis of this region identified an HGO gene locus with 14 expressed exons and 13 intervening introns (Step 3). DNA sequence analysis of multiple individuals shows a larger number of Single Nucleotide Polymorphism (SNP) variants in particular exons. Two of these SNPs are predicted to cause amino acid substitutions in the protein products of Exons 10 & 12. These are respectively a change of Pro to Ser at residue 230 (P230S), and substitution of Glu for Val at residue 300 (V300G) (Step 4). A child with Alkaptonuria is born to unaffected parents: DNA sequencing shows that the parents have the two different allelic variants (Step 5), each in combination with an alternative "+" allele. DNA sequence analysis shows that the unaffected siblings have the three possible combinations of the "+", P230S, and V300G alleles. The affected child has inherited both the P230S and V300G alleles, both of which are non-functional. HOMEWORK: what SNPs are responsible for the two amino acid substitutions?
Garrod's analysis in the early 20th century is an example of Classical or Mendelian Genetics: given observable phenotypic variation, he inferred the genotypic nature of inheritance from an analysis of pedigrees. This differs slightly from what Mendel did, which was to arrange controlled crosses and measure proportions in the observed outcomes. This is how the science of Genetics was understood for more than 50 years, which only in the molecular era was sometimes described as "Forward Genetics". Molecular Biology in the late 20th and early 21st centuries is sometimes called "Reverse Genetics", because detailed knowledge of the molecular genotype predicts how it produces a disease phenotype that arises in certain pedigrees. Confusion arises with the formulation of the Central Dogma as "DNA makes RNA makes Protein", which refers to the forward transfer of information. The logic of the inference is from detection of an enzyme defect to the underlying DNA variant, and thence to phenotype, which implies a "reversal" of molecular logic.
For the advanced student: Mendel's experiments carefully isolated and brought together exactly two allelic variants of each gene, A & a. Garrod's interpretation was that any individual with Alkaptonuria combined two copies of the same "a", such that the individual was an aa homozygote. This tacitly assumes that there are only two alleles, the "normal" A allele carried by most people, and the "disease" a allele found in affected persons. This became the standard interpretation: most individuals were homozygous AA for a standard "wild type" allele A, whereas a minority were homozygous aa for a "disease" allele a. Early molecular analysis began to show instead that genetic defects might occur at several places in the DNA of any gene. The combination of two different alleles a'a could produce a compound
heterozygote, with the genetic
phenotype of an aa homozygote.
True homozygosity would
require that an individual inherited an allele identical
by descent (autozygous)
from a more or less distant ancestor. One result of
the Human Genome Project
is to demonstrate extensive heterogeneity among the
alleles associated with any particular genetic
condition, such that compound heterozygosity
is far more frequent than
previously expected.
HOMEWORK: Step 3 of the analysis presented above (observation of a 3:1 ratio of unaffected to affected children) is an exaggeration. Garrod's actual data departed from 3:1 due to ascertainment bias. Investigate and explain.
Molecular Genetics in the 21st century proceeds from knowledge of the 3,200 Mbp human genome, completed in 2003 (Step 1). Based on knowledge of the amino acid sequences of HGO in other organisms, bioinformatic analysis of all 23 pairs of chromosomes mapped the gene for Homogentisic Acid Oxidase (HGO) to Band 2 on the long (q) arm of Chromosome 3 (3q2) (Step 2). Detailed sequence analysis of this region identified an HGO gene locus with 14 expressed exons and 13 intervening introns (Step 3). DNA sequence analysis of multiple individuals shows a larger number of Single Nucleotide Polymorphism (SNP) variants in particular exons. Two of these SNPs are predicted to cause amino acid substitutions in the protein products of Exons 10 & 12. These are respectively a change of Pro to Ser at residue 230 (P230S), and substitution of Glu for Val at residue 300 (V300G) (Step 4). A child with Alkaptonuria is born to unaffected parents: DNA sequencing shows that the parents have the two different allelic variants (Step 5), each in combination with an alternative "+" allele. DNA sequence analysis shows that the unaffected siblings have the three possible combinations of the "+", P230S, and V300G alleles. The affected child has inherited both the P230S and V300G alleles, both of which are non-functional. HOMEWORK: what SNPs are responsible for the two amino acid substitutions?
Garrod's analysis in the early 20th century is an example of Classical or Mendelian Genetics: given observable phenotypic variation, he inferred the genotypic nature of inheritance from an analysis of pedigrees. This differs slightly from what Mendel did, which was to arrange controlled crosses and measure proportions in the observed outcomes. This is how the science of Genetics was understood for more than 50 years, which only in the molecular era was sometimes described as "Forward Genetics". Molecular Biology in the late 20th and early 21st centuries is sometimes called "Reverse Genetics", because detailed knowledge of the molecular genotype predicts how it produces a disease phenotype that arises in certain pedigrees. Confusion arises with the formulation of the Central Dogma as "DNA makes RNA makes Protein", which refers to the forward transfer of information. The logic of the inference is from detection of an enzyme defect to the underlying DNA variant, and thence to phenotype, which implies a "reversal" of molecular logic.
For the advanced student: Mendel's experiments carefully isolated and brought together exactly two allelic variants of each gene, A & a. Garrod's interpretation was that any individual with Alkaptonuria combined two copies of the same "a", such that the individual was an aa homozygote. This tacitly assumes that there are only two alleles, the "normal" A allele carried by most people, and the "disease" a allele found in affected persons. This became the standard interpretation: most individuals were homozygous AA for a standard "wild type" allele A, whereas a minority were homozygous aa for a "disease" allele a. Early molecular analysis began to show instead that genetic defects might occur at several places in the DNA of any gene. The combination of two different alleles a'a could produce
HOMEWORK: Step 3 of the analysis presented above (observation of a 3:1 ratio of unaffected to affected children) is an exaggeration. Garrod's actual data departed from 3:1 due to ascertainment bias. Investigate and explain.

Archibald Garrod (ca. 1908)