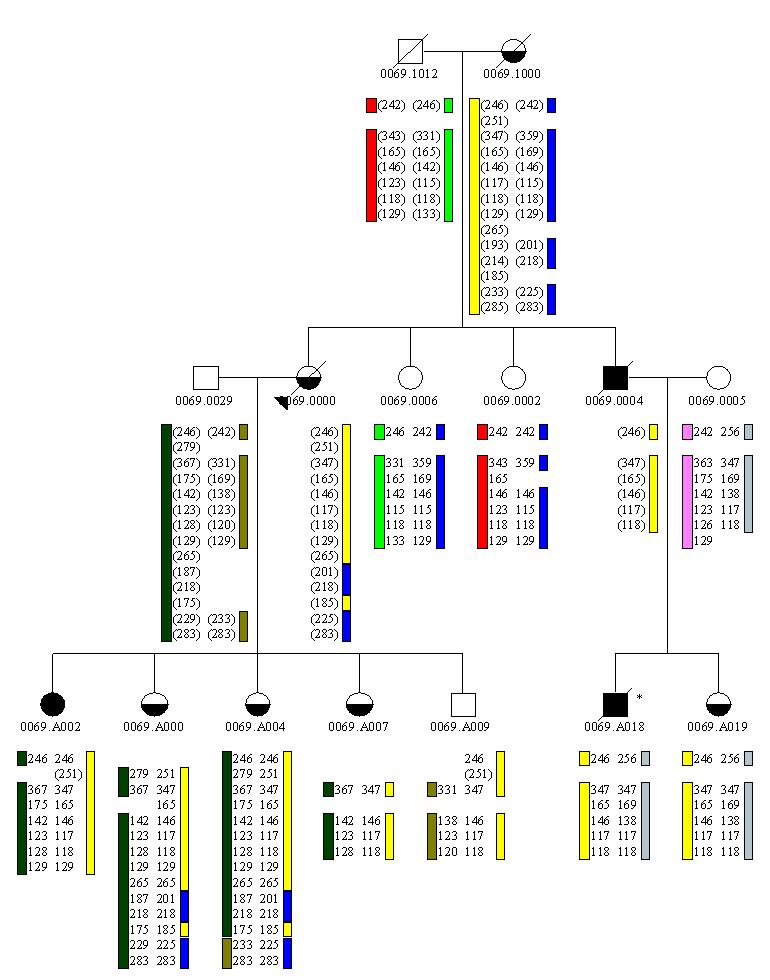
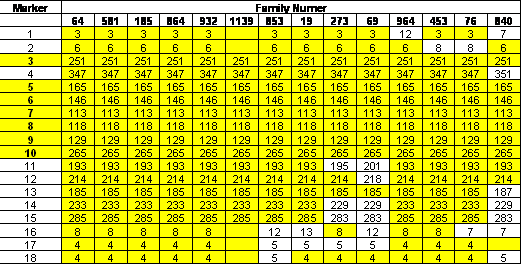
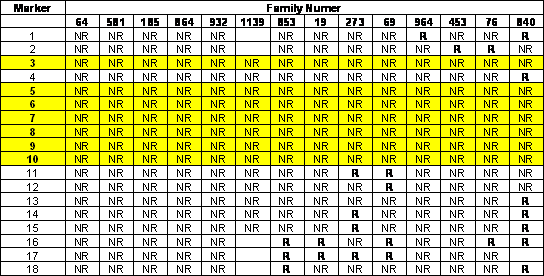
References:
1. McKenna
WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine
G, Camerini F. Diagnosis of arrhythmogenic right ventricular
dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial
and Pericardial Disease of the European Society of Cardiology and of
the Scientific Council on Cardiomyopathies of the International
Society and Federation of Cardiology. BHJ 1994; 71(3):215-218.
2.
Hodgkinson KA, Parfrey PS, Bassett AS, Kupprion C, Drenckhahn J,
Norman MW, Thierfelder L, Stuckless SN, Dicks EL, McKenna WJ,
Connors SP. The impact of implantable
cardioverter-defibrillator therapy on survival in autosomal-dominant
arrhythmogenic right ventricular cardiomyopathy (ARVD5). JACC
2005; 45(3):400-408.
3. Nava A, Scognamiglio R, Thiene G, et al.
A polymorphic form of familial arrhythmogenic right ventricular
dysplasia. Am J Cardiol 1987;59:1405–1409.
4. Ahmad F, Li D,
Karibe A, Gonzalez O, Tapscott T, Hill R, Weilbaecher D, Blackie P,
Furey M, Gardner M, Bachinski LL, Roberts R. Localization of a
gene responsible for arrhythmogenic right ventricular dysplasia to
chromosome 3p23. Circulation. 1998; 98(25):2791-2795.
5.
Gerull B, Osterziel KJ, Witt C, Dietz R, Thierfelder L. A rapid
protocol for cardiac troponin T mutation detection in familial
hypertrophic cardiomyopathy. Hum Mutat 1998;11:179–82.
6. W.
Marshall, M. Furey and B. Larsen et al., Right ventricular
cardiomyopathy and sudden death in young people. N Engl J Med 1988;
319:174–175.
7. Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch
E, Sen-Chowdhry S, McKenna WJ, Arrhythmogenic right
ventricular dysplasia/cardiomyopathy associated with mutations in
the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;
79(5):978-984.5. Gerull B, Osterziel KJ, Witt C, Dietz R,
Thierfelder L. A rapid protocol for cardiac troponin T mutation
detection in familial hypertrophic cardiomyopathy. Hum Mutat
1998;11:179–82.
| |