Transitions (Ts) are more frequent than Transversions (Tv)
[Ts = C
'silent' >> 'replacement' substitutions
3rd position >> 2nd & 1st substitutions (usually)
Molecules provide an independent estimate of phylogeny
Avoids
circular argument:
Morphology
is
used to create a classification,
then the classification is interpreted to explain
evolution
Ex.: Chinese water deer (Hydropotes)
are the only antlerless deer
=>
placed
in a separate subfamily
&
assumed
to be ancestral type
But,
molecular
analysis shows antlers were lost secondarily
Molecules provide large numbers of characters for analysis
Homo has ca. 200 bones and 3,000,000,000 nucleotide
pairs
Typical
morphological
study involves <100 characters
Typical
molecular
study involves >1,000
Patterns of molecular evolution are understood
Transitions
(Ts) are more frequent than Transversions (Tv)
[Ts = C![]() T
or A
T
or A![]() G, Tv
everything else]
G, Tv
everything else]
'silent'
>> 'replacement' substitutions
3rd
position
>> 2nd & 1st substitutions (usually)
Relative importance of characters is easier to judge
Is
the
# of toes more important than # of teeth?
Are
scales
versus feathers more important than # of temporal
openings?
But: Any one nucleotide position is more or less like any
other
1. Defining the systematic problem: "Is the Giant Panda a bear or a raccoon?"
Evolutionary
relationships of the Giant
Panda (Ailuropoda)
Ailuropoda
has been considered to be either a bear
(Ursidae) or a raccoon
(Procyonidae)
General morphology suggests ursid ancestors:
Details
of
skull, diet, biogeography suggest procyonid ancestors
Ex.: alar canal is present in Ursidae (including Ailuropoda),
absent
in Procyonidae, except lesser panda (Ailurus)
2. Collecting the data:
Measure homologous
characters in a set of taxa:
with
DNA, each nucleotide position is a separate character
mitochondrial DNA (mtDNA)
is used in many systematic studies
"Small
circular
molecule ...", 16Kbp, maternally-inherited (cytoplasmic)
13
protein loci, 2 rDNA & 22 tDNA genes (slow),
control region (very fast)
'Universal
primers' permit PCR & DNA sequencing
from many taxa
cytochrome b gene is
widely used:
Large
data
base for comparison
1140
bp
in most vertebrates
3. Analyzing the data:
Phenetic (how similar
are taxa?)
versus
cladistic (how
closely related are taxa?) criteria
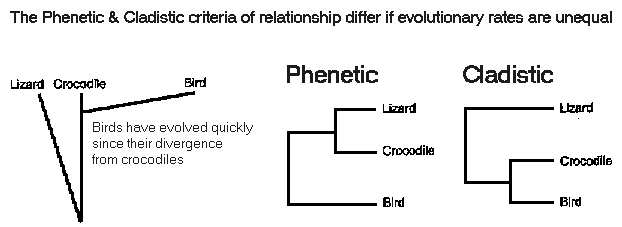
These criteria agree, iff
rates of evolution are constant
If
evolutionary rates differ, closely related organisms may appear
different
Ex.: Crocodiles
more closely related to birds, but more similar
to lizards
Crocodiles
resemble
lizards more than birds
because
birds
rapidly evolved specializations for flight
A. Phenetic analysis
Simplest measure is % sequence similarity (S)
p-distance
= (1 - S) x 100
Patterns of similarity can be inferred from cluster analysis
Most
widely
used is UPGMA
[Unweighted Pair Group Method
with
Averaging],
a Sequential Agglomerative Hierarchical Nesting (SAHN) algorithm
[algorithm = a set of
instructions for doing a repetitive task]
In (n) x (n) matrix, join the most similar pair
re-calculate (n-1) x (n-1) matrix, re-join,
and
so
on, until last pair is joined
Results are show as a phenogram:
a
diagram
of phenetic relationships
UPGMA method assumes that rates of evolution are equal
so
branch tips "come
out even" (contemporaneous)
Some
alternatives:
Neighbor-Joining (NJ) analysis
does not assume rate equality
large
evolutionary
rate differences lead to incorrect
trees
NJ
allows
branch lengths proportional to change: tips come out uneven
[algorithm
joins
nodes, rather than tips]
This
method
is more realistic, computationally harder
[see www.megasoftware.net
for free software]
Differential weighting of
nucleotide substitutions
accord
greater
'significance' to 'important' changes
Ex.: Kimura 2-parameter
distance (K2P) model treats Ts & Tv separately
K ![]() transition
bias
=
[Ts] / [Tv]
transition
bias
=
[Ts] / [Tv]
There
are
twice as many
kinds of transversions as transitions:
expected K = 0.5
But: Tv are rare for close comparisons,
more
common
for distant relationships
K is variable according to the evolutionary problem under
consideration:
K > 6 for close comparisons
B. Cladistic Analysis
Principles of homology
& analogy can be applied to nucleotide
changes
We
rely
only on shared derived (synapomorphic) nucleotide
sites,
& avoid shared ancestral (symplesiomorphic)
nucleotide sites,
and
changes
unique to single taxa (autapomorphies),
and convergent nucleotides between unrelated taxa.
Choice
of preferred hypothesis is made on the Principle of Parsimony
In
general:
parsimony means that the simpler hypothesis is to
be preferred
complex
hypotheses
are less probable
Evolutionary parsimony:
a
hypothesis
that requires fewer character changes is preferred
Ex.: to explain the origin of a complex structure
it
is
more parsimonious to hypothesize that it has evolved only once
In
molecular
systematics, these changes are nucleotide substitutions
[DNA mutations]
The "Four-Taxon
Problem" and the "Three-Taxon Statement":
Among four taxa A, B, C, & D, there are three
hypotheses of relationship:
either A is most closely related to B, or to C,
or to D
We
want
to be able to evaluate hypotheses of the form:
"X and Y are more closely
related to each other than either is to Z"
The
alternative
hypotheses can be shown as networks with branches
and an internode
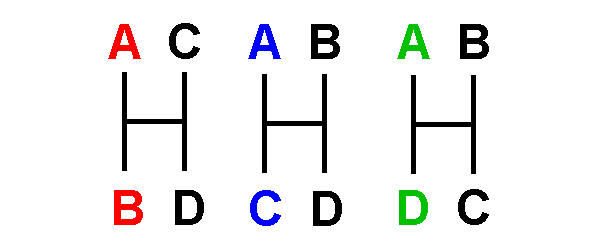

Types
1 - 4 are uninformative:
They
give
no information about relationships, because
all
hypotheses require the same number of changes,
so none is more parsimonious
than the others.
Type 1 is invariant: no changes are required.
Type 2 indicates only that one
taxon is unique wrt the
others:
all
hypotheses
require a single nucleotide change.
Type 3 indicates that all taxa
are distinct & unique:
all
hypotheses
require three nucleotide changes.
Type 4 indicates that two taxa
are similar,
but not whether this similarity is ancestral
or derived:
shared a could be either
hypothesis
requires
two changes
Alternative
hypotheses
also require two nucleotide changes.
[a '+' indicates a change along a particular network
branch]
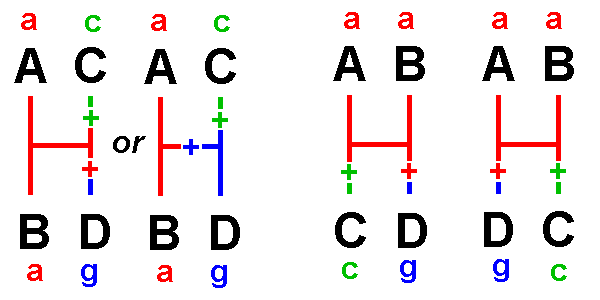
Types 5, 6
& 7 are informative:
They
give
information about relationships,
because one hypothesis requires fewer changes
than the others
&
is
therefore more parsimonious than the others
Type
5 indicates that A & B are
most closely related:
The
first
hypothesis can explain the distribution of nucleotides with a
single change,
the
latter
two require two changes each. [See also lab #5]
The
first
hypothesis
is a more parsimonious explanation of the data than the others.
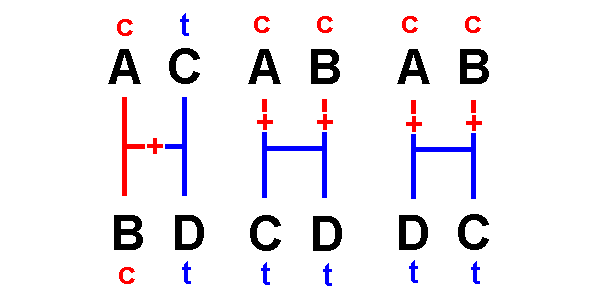
By the same logic:
Type 6 indicates
that A & C are most closely related.
Type 7 indicates
that A & D are most closely related.
[Homework:
for the three networks above,
sketch the changes required by sites of types 6 & 7]
A cladistic
analysis counts the number of
informative characters favoring each hypothesis
The
hypothesis with the "highest
score" requires the fewest changes
and
is
therefore the 'most parsimonious' explanation.
This
is
also called the 'minimum length' solution.
Cladistic
analyses may also be weighted:
Ex.:
Count
Tv:Ts as 3:1  Tv are 3x as meaningful
Tv are 3x as meaningful
or, count Tv only (Transversion
parsimony) for "deep" analyses
or, count 1st & 2nd position substitutions
>> 3rd
C. Placing the root & Inferring the direction of evolutionary change
Suppose the first
hypothesis (A & B are most closely
related) is most parsimonious
Ex.:
In Lab #5, we found that
the majority of sites were of type #5. We said:
"Ailuropoda & Ursus are
more closely related to each other
than
either
is to Procyon (or Martes)."
The
hypothesis
can be drawn as an unrooted
network
But: this evidence can also be used to argue
"Procyon & Martes are
more closely related to each other
than
either
is to Ursus (or Ailuropoda)."
To
resolve
this, we need to know where their common ancestor fits in.
There are four branches and
one internode in this network
An evolutionary tree is
a
network with a root:
The
root indicates the relationship with the common ancestor
A 'root' can be placed on any of the branches or the
internode.
So,
there
are five possible rooted
trees for this unrooted network.
All
are
equally parsimonious:
not
all
place A & B as each other's closest
relatives.
Some
of
these make shared characters symplesiomorphic
There are several ways to placement the root
(1) Outgroup rooting:
Include
a
taxon that is known to be less closely related
to
any
of the ingroup taxa than
they are to each other.
Such
a
taxon is called an outgroup or
sister taxon.
Ex.: Lynx (Feloidea)
is an outgroup to the Canoidea
(Note
that
this tree is equivalent to the NJ phenogram)
(2) Midpoint rooting:
Place
the
root halfway between
the
two
most divergent taxa.
This
assumes
that molecular evolution is clock-like.
(Here,
this
places the root on the internode).
(3) Character
Polarity:
If
the
character state of the ancestor is known (or can be inferred).
Root
the
tree accordingly
Use
of
polarity is usually not possible with molecular data
Any
nucleotide
can mutate to any other, in either direction
any a c g t looks exactly
like any other a c g t
[Some
models
allow for differential probabilities of mutation]
Homologous
nucleotide
in ancestor has most likely mutated
Use
of
polarity with morphological data is standard
Ex.: In an analysis of the evolution of the number of
heart chambers in
codfish (2), lizard (3), crocodile (4),
& bird (4)
we
know
that the evolutionary order is 2 ![]() 3
3![]() 4
4
(this
is
called a transformation series)
The
root will be placed on the codfish branch,
because
we
know the codfish most resembles the ancestor.
Crocs
&
Birds have common ancestor with a four-chambered heart.
D. Statistical tests
determine confidence in branching order
Bootstrap Analysis: a
re-sampling technique
statistical
tests
usually involve obtaining replicates / repeating experiment
Suppose
existing
data set (401bp) is a random sample of parametric data set
(complete genome)
re-sample existing n sites 1000 times,
repeat phylogenetic analysis:
how
often
do same clades / clusters appear?
"50% bootstrap support"
indicates particular group
occurs
more
frequently than all others combined
95% criterion is desirable, not often obtained with small data sets
What does this analysis explain about the biology & evolution of Pandas?
1. Ailuropoda and Ursus are each
others' closest
relatives:
The
Giant
Panda is a highly derived bear, not a
raccoon.
Ailuropoda should be classified in Ursidae.
2. Similarities
of Ailuropoda
and Ailurus are convergent
(analogous):
these
represent
parallel feeding specializations.
Ex.: "Hypertrophied masticatory apparatus" permits
feeding on bamboo:
(expanded zygomatic arch,
high mandibular ramus, and molariform teeth)
Jaw
articulation
above toothrow gives mechanical advantage:
(similar
modifications
occur in Hyaena for crushing bones).
3.
Some similarities between Ailuropoda and other
ursids are ancestral homologies:
Bears
(including
pandas) have short gestation and tiny neonates.
In
most
bears, gestation & birth occur during winter hibernation:
Hypothesis: early
parturition (birth) gives access to milk, when no
other food is available
But: Pandas do not hibernate, young are carried during
foraging:
Why
have
altricial (underdeveloped) young when food is readily
available?
| "Small young could be explained if the suite of physiological and behavioural adaptations associated with the production of small neonates were established before splitting of the panda and ursid lines." (Ramsay & Dunbrack, 1987) |
That is, tiny neonates are a conserved ancestral condition rather than a a contemporary adaptive response.
4. "Evo-Devo"
basis of Panda
evolution
Development
& growth of cranial versus axial skeleton in pandas
resembles Hyaenas and boxer dogs:
Heavy
crania, less-developed post-cranial (axial) skeletons.
Selection
may operate on similar, hypothetical 'growth fields'
Text material © 2020 by Steven M. Carr
{kind=link}